05_plot_the_results
[1]:
from pigeon_feather.data import *
from pigeon_feather.plot import *
from pigeon_feather.hxio import *
from pigeon_feather.spectra import *
import numpy as np
import pandas as pd
import datetime
import os
import pickle
import datetime
Determination of memory status is not supported on this
platform, measuring for memoryleaks will never fail
[2]:
# load the pickle file we saved in the previous notebook
today = datetime.date.today().strftime("%Y%m%d")
today = "20240722"
with open(f"./data/hdxms_data_raw_{today}.pkl", "rb") as f:
hdxms_data_list = pickle.load(f)
# back exchange correction for peptides with experimental full deuteration data based its closest match in the database
tools.backexchange_correction(hdxms_data_list)
Number of peptides with experimental max_d: 358
Number of peptides with no experimental max_d: 12
[3]:
# make folders for results
today_date = datetime.date.today().strftime("%Y%m%d")
# today_date = '20240722'
results_path = f"ecDHFR_results_{today_date}"
if not os.path.exists(results_path):
os.makedirs(results_path)
out_path = "./data/PF_input_20240722"
Create an Analysis object and load the results. Note: The temperature and pH are crucial for calculating the intrinsic exchange rates, so make sure to input the correct values. The chunk size and chunk number can be altered by the user. In this tutorial, they were automatically determined by a helper function.
[4]:
from pigeon_feather.analysis import Analysis, get_index_offset
from pigeon_feather.tools import optimal_chunks
import matplotlib.pyplot as plt
RUN_NUM = 3
apo_states = [
state
for data in hdxms_data_list
for state in data.states
if state.state_name == "APO"
]
tri_states = [
state
for data in hdxms_data_list
for state in data.states
if state.state_name == "TRI"
]
ana_apo_1 = Analysis(apo_states, temperature=293.0, pH=7.0)
chunk_num, chunk_size = optimal_chunks(len(ana_apo_1.protein_sequence))
ana_apo_1.load_bayesian_hdx_oupt_chunks(
chunk_size=chunk_size,
chunk_num=chunk_num,
state_name="APO",
run_num=RUN_NUM,
N=200,
bayesian_hdx_data_folder=f"{out_path}/bayesian_hdx_output_chunks",
)
ana_tri_1 = Analysis(tri_states, temperature=293.0, pH=7.0)
ana_tri_1.load_bayesian_hdx_oupt_chunks(
chunk_size=chunk_size,
chunk_num=chunk_num,
state_name="TRI",
run_num=RUN_NUM,
N=200,
bayesian_hdx_data_folder=f"{out_path}/bayesian_hdx_output_chunks",
)
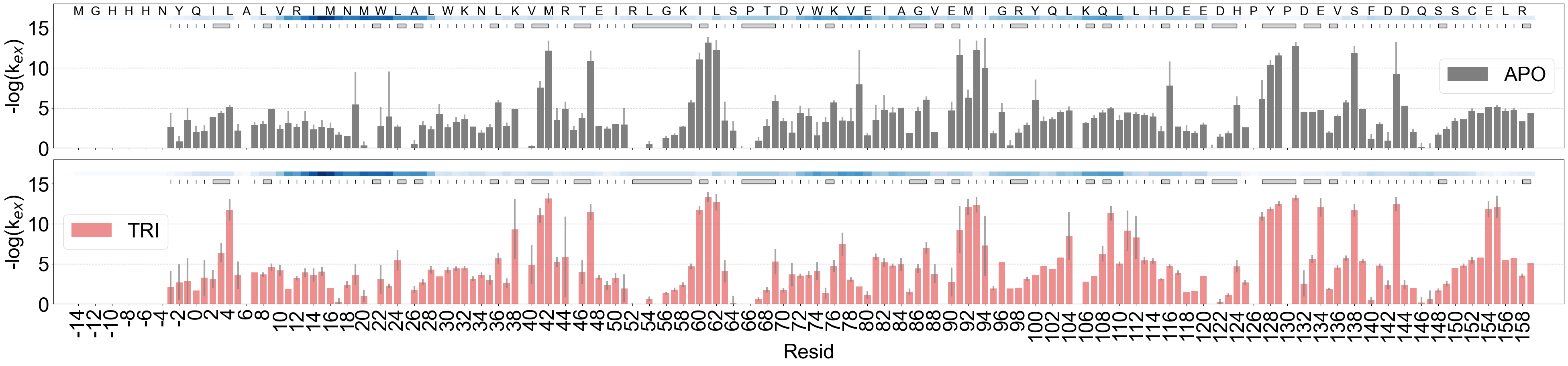
barplot of the kex
barplot showing the FEATHER derived exchange rates. index_offset is determined by comparison to the sequence in HX/MS and the pdb file provided.
[5]:
# APO state only
import numpy as np
fig, axes = plt.subplots(2, 1, figsize=(40, 10), sharey=True, sharex=True)
ana_apo_1.plot_kex_bar(
ax=axes[0], resolution_indicator_pos=15, label="APO", show_seq=False, color="k"
)
ana_tri_1.plot_kex_bar(
ax=axes[1], resolution_indicator_pos=15, label="TRI", show_seq=False, color="red"
)
from matplotlib.pyplot import step
from matplotlib.ticker import FuncFormatter, MultipleLocator
from pigeon_feather.analysis import get_index_offset
pdb_file = "./data/6XG5_TRI.pdb"
index_offset = get_index_offset(ana_apo_1, pdb_file)
# # ax.xaxis.set_major_locator(plt.MultipleLocator(2))
axes[0].set_xticks(axes[0].get_xticks()[::2])
axes[0].set_xticklabels(axes[0].get_xticklabels(), fontdict={"fontsize": 24})
def format_func(value, tick_number):
return f"{int(value - index_offset +1)}"
axes[0].xaxis.set_major_formatter(FuncFormatter(format_func))
seq_pos = 17
for ii in range(0, len(ana_apo_1.protein_sequence[:]), 2):
axes[0].text(
ii,
seq_pos,
ana_apo_1.protein_sequence[ii],
ha="center",
va="center",
fontsize=22,
)
for ax in axes:
ax.set_ylabel("-log(k$_{ex}$)")
#ax.legend(loc="lower left")
ax.yaxis.set_major_locator(plt.MultipleLocator(5))
ax.plot(
[-3, len(ana_apo_1.protein_sequence) + 3],
[5, 5],
color="gray",
lw=1,
linestyle="--",
alpha=0.5,
)
ax.plot(
[-3, len(ana_apo_1.protein_sequence) + 3],
[10, 10],
color="gray",
lw=1,
linestyle="--",
alpha=0.5,
)
plt.subplots_adjust(hspace=0.08)
fig.savefig(f"{results_path}/ecDHFR_kex_bar_APO_TRI_{today_date}.pdf")
logP projection to a pdb structure
[6]:
from pigeon_feather.analysis import BFactorPlot
[7]:
bfactor_plot = BFactorPlot(
ana_apo_1,
pdb_file="./data/6XG5_TRI.pdb",
plot_deltaG=True,
temperature=293.15,
logP_threshold=10,
)
bfactor_plot.plot(f"{results_path}/6XG5_APO_deltaG.pdb")
# bfactor_plot = BFactorPlot(
# ana_apo_1,
# pdb_file="./data/6XG5_APO_relaxed_best_solvated.pdb",
# plot_deltaG=True,
# temperature=293.15,
# )
# bfactor_plot.plot(f"{out_path}/6XG5_APO_deltaG.pdb")
delta log(PF)s
if there are mutiple states availble, you can map the difference to the pdb strutcure
[8]:
bfactor_plot = BFactorPlot(
ana_apo_1,
ana_tri_1,
pdb_file="./data/6XG5_TRI.pdb",
plot_deltaG=True,
temperature=293.15,
logP_threshold=10,
)
bfactor_plot.plot(f"{results_path}/6XG5_deltaG_APO-TRI.pdb")
You can visualize the results in PyMOL by running the following commands:
spectrum b, green_white_magenta, minimum=-20, maximum=20; select nans, not (b=0 or b>0 or b<0); color grey20, nans;
nans are Prolines and residues with no coverage.
export logPFs to a csv
[9]:
import MDAnalysis
from pigeon_feather.analysis import get_res_avg_logP, get_res_avg_logP_std
pdb_file = "./data/6XG5_TRI.pdb"
index_offset = get_index_offset(ana_apo_1, pdb_file)
def logPF_to_deltaG(ana_obj, logPF):
"""
:param logPF: logP value
:return: deltaG in kJ/mol, local unfolding energy
"""
return 8.3145 * ana_obj.temperature * np.log(10) * logPF / 1000
def create_logP_df(ana_obj, index_offset):
df_logPF = pd.DataFrame()
for res_i, _ in enumerate(ana_obj.results_obj.protein_sequence):
res_obj_i = ana_obj.results_obj.get_residue_by_resindex(res_i)
avg_logP, std_logP = get_res_avg_logP(res_obj_i)
#std_logP = get_res_avg_logP_std(res_obj_i)
df_i = pd.DataFrame(
{
"resid": [res_obj_i.resid - index_offset],
#"resname": [res_obj_i.resname],
"resname": [MDAnalysis.lib.util.convert_aa_code(res_obj_i.resname)],
'avg_dG': [logPF_to_deltaG(ana_obj, avg_logP)],
'std_dG': [logPF_to_deltaG(ana_obj, std_logP)],
"avg_logP": [avg_logP],
"std_logP": [max(std_logP, 0.35)],
"is_nan": [res_obj_i.is_nan()],
}
)
if res_obj_i.is_nan():
df_i["single resloved"] = [np.nan]
df_i["min_pep logPs"] = [np.nan]
else:
df_i["single resloved"] = [res_obj_i.mini_pep.if_single_residue()]
df_i["min_pep logPs"] = [res_obj_i.clustering_results_logP]
df_logPF = pd.concat([df_logPF, df_i])
df_logPF = df_logPF.reset_index(drop=True)
return df_logPF
df = create_logP_df(ana_apo_1, index_offset)
df.to_csv(f"{results_path}/logP_APO_{today_date}.csv", index=False)
[10]:
# fecth two df and merge
df_apo = create_logP_df(ana_apo_1, index_offset)
df_tri = create_logP_df(ana_tri_1, index_offset)
df = pd.merge(df_apo, df_tri, on="resid", suffixes=("_APO", "_TRI"))
df.to_csv(f"{results_path}/logP_APO_TRI_{today_date}.csv", index=False)
[ ]: